[ Home Page ] [ Site Map ]
Acute Lymphoblastic Leukaemia
Clinical Features
Laboratory Diagnosis
Cytogenetics
Prognostic Factors
Outcome and Therapy
Acute lymphoblastic leukaemia (ALL) can occur at any age but has a peak incidence between 2 - 10 years. It is characterised by:
- bone marrow failure
- lymphadenopathy
- thymic enlargement in T-lineage ALL
- bone pain which may be associated with radiological change and fracture
- tendency to relapse in the CNS and testis.
Morphology
[Ref 1]
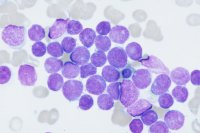
Blast morphology is variable - some are small with high nuclear:cytoplasmic ratios and indistinct nucleoli (so-called L1 blasts) while others are larger with more prominent nucleoli and more abundant cytoplasm (L2). Blast morphology does not correlate with cell lineage and cytochemistry is of little value.
 |
|
ALL. Bone marrow. Complete replacement by small/medium sized blasts with scanty cytoplasm and round nuclei with dense chromatin (FAB L1 type, common-ALL phenotype). |
ALL. Bone marrow. Pleiomorphic blasts with variable amounts of cytoplasm, twisted irregular nuclei and multiple indistinct nucleoli. (FAB L2 type, common-ALL phenotype). |
|
|
Burkitt lymphoma, leukaemic phase. Bone marrow. Deeply basophilic blasts with dense nuclear chromatin and multiple cytoplasmic vacuoles. t(8;14) present. |
ALL. Peripheral blood. Large blasts with convoluted nuclei and basophilic cytoplasm. (T-ALL phenotype). |
Immunophenotype
[Ref 1]
ALL is derived from precursor lymphocytes that are undergoing antigen receptor gene (Ig and TCR) rearrangement.
B-lineage ALL
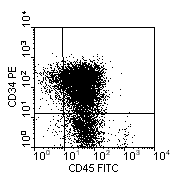
The precursor nature of the cells is established by demonstrating lack of surface Ig, the presence of nuclear Tdt and sometimes the expression of CD34. Subclassification is as follows:
- Pre-pre B-ALL: CD19+ CD10- cytoplasmic mu heavy chain negative.
- Common ALL: CD19+ CD10+ cytoplasmic mu present in <20% of cells
- Pre B-ALL: CD19+ CD10+ cytoplasmic mu present in >20% of cells
- Blasts of all subgroups will express cytoplasmic CD22 and CD79b.
 |
 |
 |
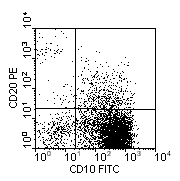
Two-colour immunophenotypic profile of common-ALL: blast cells positive for CD34 and CD10. |
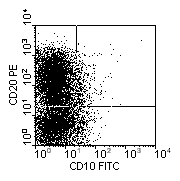
Pre pre-B ALL blast cells lack expression of CD10. |
T-lineage ALL
The precursor nature of the cells is established by demonstrating Tdt and sometimes CD34 positivity and the lack of surface TCR/CD3. T-cell lineage is demonstrated by the expression of CD7 and/or CD1a. Expression of the other pan-T cell markers is variable.
Hyperdiploidy is common[2].
A number of balanced translocations have been identified in ALL:
- t(12;21) - this is the commonest translocation in ALL (30% of cases). It results in the TEL-AML fusion gene and is primarily associated with the common phenotype[3].
- t(9;22) - this is commoner in adults and is associated with a very poor prognosis[2,4].
- t(4;11) - this translocation results in the MLL-AF4 fusion gene. It is associated with pre-pre B-ALL and is associated with a poor prognosis[5,6].
- t(1;19) - associated with pre-B ALL and results in the formation of the E2A-PBX fusion gene[7,8].
These translocations are demonstrable by RT-PCR techniques.
TAL-1 deregulation is the commonest genetic abnormality in T-ALL. This may occur as the result of the t(1;14) or more commonly due to chromosome 1p32 deletions[9].
The following are poor prognostic factors in ALL:
- age <1 year and >10 years
- male sex
- CNS disease at presentation
- high white cell count
- t(9;22)
- t(4;11)
- hypodiploidy
The treatment of ALL consists of the following "phases":
- Remission induction - vincristine, prednisolone, daunorubicin, asparaginase.
- Consolidation - various combinations of chemotherapeutic agents.
- CNS directed therapy - high dose systemic and intrathecal methotrexate.
- Maintenance therapy - vincristine, prednisolone, mercaptopurine and methotrexate for 2 years.
Childhood ALL is associated with 75% long term survival. Minimal residual disease assessment using PCR based strategies appear to be able to predict relapse although they are not yet in routine clinical use[10]. Allogeneic transplantation is the treatment of choice at relapse.
The outlook in adult ALL is poor with approximately 20% long-term survivors. Allogeneic transplantation is advisable in first remission.

[ Home Page ] [ Site Map ]
Copyright © HMDS 1999-2005
Document last updated:
Tuesday, 18 November 2003
Comments & feedback to:
admin@hmds.org.uk
[URL: www.hmds.org.uk/all.html]